VAE를 사용한 약물 분자 생성
- 원본 링크 : https://keras.io/examples/generative/molecule_generation/
- 최종 확인 : 2024-11-23
저자 : Victor Basu
생성일 : 2022/03/10
최종 편집일 : 2022/03/24
설명 : 약물 발견을 위한 컨볼루션 변분 오토인코더 (VAE, Variational AutoEncoder) 구현
소개
이 예시에서는, Variational Autoencoder (VAE)를 사용하여 약물 발견을 위한 분자를 생성합니다. 이 예시는 논문 Automatic chemical design using a data-driven continuous representation of molecules와 MolGAN: An implicit generative model for small molecular graphs를 참조하였습니다.
논문 Automatic chemical design using a data-driven continuous representation of molecules에 설명된 모델은, 화합물의 열린 공간(open-ended spaces)을 효율적으로 탐색하여 새로운 분자를 생성합니다. 이 모델은 인코더(Encoder), 디코더(Decoder) 및 예측기(Predictor)의 세 가지 구성 요소로 이루어져 있습니다. 인코더는 분자의 이산 표현을 실수로 된 연속 벡터로 변환하고, 디코더는 이러한 연속 벡터를 다시 이산적인 분자 표현으로 변환합니다. 예측기는 분자의 연속적인 벡터 표현에서 화학적 특성을 추정합니다. 연속적인 표현은 기울기 기반 최적화를 사용하여 최적화된 기능성 화합물을 효율적으로 탐색할 수 있게 합니다.
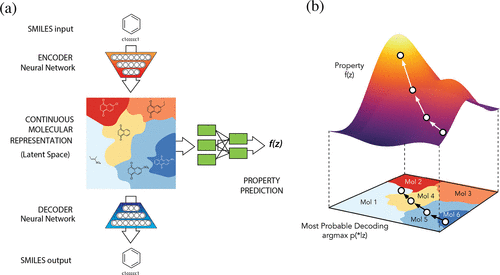
그림 (a)
- 분자 설계를 위한 오토인코더 다이어그램으로, 공동 특성(joint property) 예측 모델을 포함합니다.
- SMILES 문자열과 같은 이산적인 분자 표현을 시작으로, 인코더 네트워크는 각 분자를 잠재 공간의 벡터로 변환합니다.
- 이 벡터는 실질적으로 연속적인 분자 표현입니다.
- 잠재 공간의 한 지점을 주면, 디코더 네트워크는 이에 해당하는 SMILES 문자열을 생성합니다.
- 다중 레이어 퍼셉트론 네트워크는 각 분자와 연관된 목표 특성의 값을 추정합니다.
그림 (b)
- 연속적인 잠재 공간에서 기울기 기반 최적화입니다.
- 잠재 표현
z에 기반한 분자의 특성을 예측하기 위해 서브게이트 모델f(z)을 트레이닝한 후, - 우리는
f(z)를z에 대해 최적화하여 특정한 목표 특성에 부합할 것으로 예상되는 새로운 잠재 표현을 찾을 수 있습니다. - 이러한 새로운 잠재 표현은 SMILES 문자열로 디코딩된 후, 이들의 특성이 실험적으로 테스트될 수 있습니다.
MolGAN의 설명과 구현에 대해서는 Keras 예시 WGAN-GP with R-GCN for the generation of small molecular graphs을 참조하십시오. 현재 예시에서 사용된 많은 함수는 위의 Keras 예시에서 가져왔습니다.
셋업
RDKit는 화학정보학 및 기계 학습을 위한 오픈 소스 툴킷입니다. 이 툴킷은 약물 발견 분야에서 특히 유용합니다. 이 예시에서는, RDKit를 사용하여 SMILES를 분자 객체로 편리하고 효율적으로 변환한 후, 이 분자 객체에서 원자와 결합의 집합을 얻습니다.
WGAN-GP with R-GCN for the generation of small molecular graphs에서 인용하자면:
“SMILES는 주어진 분자의 구조를 ASCII 문자열의 형태로 표현합니다. SMILES 문자열은 작은 분자의 경우, 비교적 사람이 읽기 쉬운 압축된 인코딩입니다. 분자를 문자열로 인코딩함으로써, 주어진 분자의 데이터베이스 및/또는 웹 검색을 용이하게 합니다. RDKit는 주어진 SMILES를 정확하게 분자 객체로 변환하는 알고리즘을 사용하며, 이를 통해 수많은 분자 특성/특징을 계산할 수 있습니다.”
!pip -q install rdkit-pypi==2021.9.4import ast
import pandas as pd
import numpy as np
import tensorflow as tf
from tensorflow import keras
from tensorflow.keras import layers
import matplotlib.pyplot as plt
from rdkit import Chem, RDLogger
from rdkit.Chem import BondType
from rdkit.Chem.Draw import MolsToGridImage
RDLogger.DisableLog("rdApp.*")결과
[K |████████████████████████████████| 20.6 MB 1.2 MB/s
[?25h데이터세트
우리는 ZINC – A Free Database of Commercially Available Compounds for Virtual Screening 데이터세트를 사용합니다. 이 데이터셋에는 SMILES 표기로 표현된 분자 공식과 함께, logP (물-옥탄올 분배 계수), SAS (합성 용이성 점수), QED (약물 유사성의 정성적 추정)과 같은 해당 분자의 분자 특성이 포함되어 있습니다.
csv_path = keras.utils.get_file(
"/content/250k_rndm_zinc_drugs_clean_3.csv",
"https://raw.githubusercontent.com/aspuru-guzik-group/chemical_vae/master/models/zinc_properties/250k_rndm_zinc_drugs_clean_3.csv",
)
df = pd.read_csv("/content/250k_rndm_zinc_drugs_clean_3.csv")
df["smiles"] = df["smiles"].apply(lambda s: s.replace("\n", ""))
df.head()결과
Downloading data from https://raw.githubusercontent.com/aspuru-guzik-group/chemical_vae/master/models/zinc_properties/250k_rndm_zinc_drugs_clean_3.csv
22606589/22606589 [==============================] - 0s 0us/step| smiles | logP | qed | SAS | |
|---|---|---|---|---|
| 0 | CC(C)(C)c1ccc2occ(CC(=O)Nc3ccccc3F)c2c1 | 5.05060 | 0.702012 | 2.084095 |
| 1 | C[C@@H]1CC(Nc2cncc(-c3nncn3C)c2)C[C@@H](C)C1 | 3.11370 | 0.928975 | 3.432004 |
| 2 | N#Cc1ccc(-c2ccc(O[C@@H](C(=O)N3CCCC3)c3ccccc3)… | 4.96778 | 0.599682 | 2.470633 |
| 3 | CCOC(=O)[C@@H]1CCCN(C(=O)c2nc(-c3ccc(C)cc3)n3c… | 4.00022 | 0.690944 | 2.822753 |
| 4 | N#CC1=C(SCC(=O)Nc2cccc(Cl)c2)N=C([O-])[C@H](C#… | 3.60956 | 0.789027 | 4.035182 |
하이퍼파라미터
SMILE_CHARSET = '["C", "B", "F", "I", "H", "O", "N", "S", "P", "Cl", "Br"]'
bond_mapping = {"SINGLE": 0, "DOUBLE": 1, "TRIPLE": 2, "AROMATIC": 3}
bond_mapping.update(
{0: BondType.SINGLE, 1: BondType.DOUBLE, 2: BondType.TRIPLE, 3: BondType.AROMATIC}
)
SMILE_CHARSET = ast.literal_eval(SMILE_CHARSET)
MAX_MOLSIZE = max(df["smiles"].str.len())
SMILE_to_index = dict((c, i) for i, c in enumerate(SMILE_CHARSET))
index_to_SMILE = dict((i, c) for i, c in enumerate(SMILE_CHARSET))
atom_mapping = dict(SMILE_to_index)
atom_mapping.update(index_to_SMILE)
BATCH_SIZE = 100
EPOCHS = 10
VAE_LR = 5e-4
NUM_ATOMS = 120 # 최대 원자 수
ATOM_DIM = len(SMILE_CHARSET) # 원자 타입 수
BOND_DIM = 4 + 1 # 결합 타입 수
LATENT_DIM = 435 # 잠재 공간 크기
def smiles_to_graph(smiles):
# SMILES를 분자 객체로 변환
molecule = Chem.MolFromSmiles(smiles)
# 인접 행렬과 특성 텐서 초기화
adjacency = np.zeros((BOND_DIM, NUM_ATOMS, NUM_ATOMS), "float32")
features = np.zeros((NUM_ATOMS, ATOM_DIM), "float32")
# 분자의 각 원자에 대해 루프 실행
for atom in molecule.GetAtoms():
i = atom.GetIdx()
atom_type = atom_mapping[atom.GetSymbol()]
features[i] = np.eye(ATOM_DIM)[atom_type]
# 원-홉 이웃에 대해 루프 실행
for neighbor in atom.GetNeighbors():
j = neighbor.GetIdx()
bond = molecule.GetBondBetweenAtoms(i, j)
bond_type_idx = bond_mapping[bond.GetBondType().name]
adjacency[bond_type_idx, [i, j], [j, i]] = 1
# 결합이 없는 경우, 마지막 채널에 1을 추가하여 "비결합"을 나타냅니다.
# Notice: channels-first
adjacency[-1, np.sum(adjacency, axis=0) == 0] = 1
# 원자가 없는 경우, 마지막 열에 1을 추가하여 "비원자"를 나타냅니다
features[np.where(np.sum(features, axis=1) == 0)[0], -1] = 1
return adjacency, features
def graph_to_molecule(graph):
# 그래프 언팩
adjacency, features = graph
# RWMol은 수정 가능한 분자 객체입니다
molecule = Chem.RWMol()
# "비원자" 및 결합이 없는 원자를 제거
keep_idx = np.where(
(np.argmax(features, axis=1) != ATOM_DIM - 1)
& (np.sum(adjacency[:-1], axis=(0, 1)) != 0)
)[0]
features = features[keep_idx]
adjacency = adjacency[:, keep_idx, :][:, :, keep_idx]
# 분자에 원자 추가
for atom_type_idx in np.argmax(features, axis=1):
atom = Chem.Atom(atom_mapping[atom_type_idx])
_ = molecule.AddAtom(atom)
# 분자 내 원자 간 결합 추가; [symmetric] 인 인접 텐서의 상삼각형을 기반으로 함
(bonds_ij, atoms_i, atoms_j) = np.where(np.triu(adjacency) == 1)
for (bond_ij, atom_i, atom_j) in zip(bonds_ij, atoms_i, atoms_j):
if atom_i == atom_j or bond_ij == BOND_DIM - 1:
continue
bond_type = bond_mapping[bond_ij]
molecule.AddBond(int(atom_i), int(atom_j), bond_type)
# 분자 정화; 정화에 대한 자세한 내용은
# https://www.rdkit.org/docs/RDKit_Book.html#molecular-sanitization 을 참조하세요
flag = Chem.SanitizeMol(molecule, catchErrors=True)
# 엄격하게 처리. 정화가 실패하면 None을 반환
if flag != Chem.SanitizeFlags.SANITIZE_NONE:
return None
return molecule트레이닝 세트 생성
train_df = df.sample(frac=0.75, random_state=42) # random state는 시드 값입니다
train_df.reset_index(drop=True, inplace=True)
adjacency_tensor, feature_tensor, qed_tensor = [], [], []
for idx in range(8000):
adjacency, features = smiles_to_graph(train_df.loc[idx]["smiles"])
qed = train_df.loc[idx]["qed"]
adjacency_tensor.append(adjacency)
feature_tensor.append(features)
qed_tensor.append(qed)
adjacency_tensor = np.array(adjacency_tensor)
feature_tensor = np.array(feature_tensor)
qed_tensor = np.array(qed_tensor)
class RelationalGraphConvLayer(keras.layers.Layer):
def __init__(
self,
units=128,
activation="relu",
use_bias=False,
kernel_initializer="glorot_uniform",
bias_initializer="zeros",
kernel_regularizer=None,
bias_regularizer=None,
**kwargs
):
super().__init__(**kwargs)
self.units = units
self.activation = keras.activations.get(activation)
self.use_bias = use_bias
self.kernel_initializer = keras.initializers.get(kernel_initializer)
self.bias_initializer = keras.initializers.get(bias_initializer)
self.kernel_regularizer = keras.regularizers.get(kernel_regularizer)
self.bias_regularizer = keras.regularizers.get(bias_regularizer)
def build(self, input_shape):
bond_dim = input_shape[0][1]
atom_dim = input_shape[1][2]
self.kernel = self.add_weight(
shape=(bond_dim, atom_dim, self.units),
initializer=self.kernel_initializer,
regularizer=self.kernel_regularizer,
trainable=True,
name="W",
dtype=tf.float32,
)
if self.use_bias:
self.bias = self.add_weight(
shape=(bond_dim, 1, self.units),
initializer=self.bias_initializer,
regularizer=self.bias_regularizer,
trainable=True,
name="b",
dtype=tf.float32,
)
self.built = True
def call(self, inputs, training=False):
adjacency, features = inputs
# 이웃으로부터 정보를 집계합니다
x = tf.matmul(adjacency, features[:, None, :, :])
# 선형 변환을 적용합니다
x = tf.matmul(x, self.kernel)
if self.use_bias:
x += self.bias
# 결합 타입 차원을 줄입니다
x_reduced = tf.reduce_sum(x, axis=1)
# 비선형 변환을 적용합니다
return self.activation(x_reduced)인코더 및 디코더 빌드
인코더는 분자의 그래프 인접 행렬(adjacency matrix)과 특성 행렬을 입력으로 받습니다.
이 특성들은 그래프 컨볼루션 레이어를 통해 처리된 후,
플래튼(flatten)되고 여러 Dense 레이어를 통해,
z_mean과 log_var, 즉 분자의 잠재 공간 표현으로 변환됩니다.
그래프 컨볼루션 레이어: 관계적 그래프 컨볼루션 레이어는 비선형 변환된 이웃 집계를 구현합니다. 이를 다음과 같이 정의할 수 있습니다:
$$ H^{l+1} = σ(D^{-1} @ A @ H^{l+1} @ W^{l}) $$
여기서 $\sigma$는 비선형 변환(주로 ReLU 활성화 함수)을 나타내고,
$A$는 인접 텐서, $H^{l}$는 l번째 레이어의 특성 텐서,
$D^{-1}$는 $A^$의 역 대각(inverse diagonal) 행렬,
$W^{l}$는 l번째 레이어에서 트레이닝 가능한 가중치 텐서를 나타냅니다.
특히, 각 결합 유형(관계)마다 대각(diagonal) 행렬은 각 원자에 연결된 결합 수를 표현합니다.
출처: WGAN-GP with R-GCN을 이용한 소분자 그래프 생성
디코더는 잠재 공간 표현을 입력으로 받아, 해당 분자의 그래프 인접 행렬과 특성 행렬을 예측합니다.
def get_encoder(
gconv_units, latent_dim, adjacency_shape, feature_shape, dense_units, dropout_rate
):
adjacency = keras.layers.Input(shape=adjacency_shape)
features = keras.layers.Input(shape=feature_shape)
# 하나 이상의 그래프 컨볼루션 레이어를 통해 전파
features_transformed = features
for units in gconv_units:
features_transformed = RelationalGraphConvLayer(units)(
[adjacency, features_transformed]
)
# 2D 분자 표현을 1D로 축소
x = keras.layers.GlobalAveragePooling1D()(features_transformed)
# 하나 이상의 밀집 레이어를 통해 전파
for units in dense_units:
x = layers.Dense(units, activation="relu")(x)
x = layers.Dropout(dropout_rate)(x)
z_mean = layers.Dense(latent_dim, dtype="float32", name="z_mean")(x)
log_var = layers.Dense(latent_dim, dtype="float32", name="log_var")(x)
encoder = keras.Model([adjacency, features], [z_mean, log_var], name="encoder")
return encoder
def get_decoder(dense_units, dropout_rate, latent_dim, adjacency_shape, feature_shape):
latent_inputs = keras.Input(shape=(latent_dim,))
x = latent_inputs
for units in dense_units:
x = keras.layers.Dense(units, activation="tanh")(x)
x = keras.layers.Dropout(dropout_rate)(x)
# 이전 레이어 출력(x)을 [연속적인] 인접 텐서(x_adjacency)로 매핑
x_adjacency = keras.layers.Dense(tf.math.reduce_prod(adjacency_shape))(x)
x_adjacency = keras.layers.Reshape(adjacency_shape)(x_adjacency)
# 마지막 두 차원을 대칭화
x_adjacency = (x_adjacency + tf.transpose(x_adjacency, (0, 1, 3, 2))) / 2
x_adjacency = keras.layers.Softmax(axis=1)(x_adjacency)
# 이전 레이어 출력(x)을 [연속적인] 특성 텐서(x_features)로 매핑
x_features = keras.layers.Dense(tf.math.reduce_prod(feature_shape))(x)
x_features = keras.layers.Reshape(feature_shape)(x_features)
x_features = keras.layers.Softmax(axis=2)(x_features)
decoder = keras.Model(
latent_inputs, outputs=[x_adjacency, x_features], name="decoder"
)
return decoder샘플링 레이어 빌드
class Sampling(layers.Layer):
def call(self, inputs):
z_mean, z_log_var = inputs
batch = tf.shape(z_log_var)[0]
dim = tf.shape(z_log_var)[1]
epsilon = tf.keras.backend.random_normal(shape=(batch, dim))
return z_mean + tf.exp(0.5 * z_log_var) * epsilonVAE 빌드
이 모델은 다음 네 가지 손실을 최적화하도록 트레이닝됩니다:
- 범주형 교차 엔트로피
- KL 발산 손실
- 속성 예측 손실
- 그래프 손실(기울기 패널티)
범주형 교차 엔트로피 손실 함수는 모델의 재구성 정확도를 측정합니다. 속성 예측 손실은 잠재 표현을 속성 예측 모델에 통과시킨 후, 예측된 속성과 실제 속성 간의 평균 제곱 오차를 추정합니다. 모델의 속성 예측은 이진 교차 엔트로피를 통해 최적화됩니다. 그래프 손실은 모델의 속성(QED) 예측에 의해 추가적으로 안내됩니다.
기울기 패널티는 원본신경망에서 사용된 기울기 클리핑 방식에 대한 개선으로, 1-Lipschitz 연속성에 대한 대안적인 부드러운 제약 조건입니다. (“1-Lipschitz 연속성"은 함수의 모든 점에서 기울기의 노름(길이)이 최대 1임을 의미합니다.) 이는 손실 함수에 정규화 항을 추가합니다.
class MoleculeGenerator(keras.Model):
def __init__(self, encoder, decoder, max_len, **kwargs):
super().__init__(**kwargs)
self.encoder = encoder
self.decoder = decoder
self.property_prediction_layer = layers.Dense(1)
self.max_len = max_len
self.train_total_loss_tracker = keras.metrics.Mean(name="train_total_loss")
self.val_total_loss_tracker = keras.metrics.Mean(name="val_total_loss")
def train_step(self, data):
adjacency_tensor, feature_tensor, qed_tensor = data[0]
graph_real = [adjacency_tensor, feature_tensor]
self.batch_size = tf.shape(qed_tensor)[0]
with tf.GradientTape() as tape:
z_mean, z_log_var, qed_pred, gen_adjacency, gen_features = self(
graph_real, training=True
)
graph_generated = [gen_adjacency, gen_features]
total_loss = self._compute_loss(
z_log_var, z_mean, qed_tensor, qed_pred, graph_real, graph_generated
)
grads = tape.gradient(total_loss, self.trainable_weights)
self.optimizer.apply_gradients(zip(grads, self.trainable_weights))
self.train_total_loss_tracker.update_state(total_loss)
return {"loss": self.train_total_loss_tracker.result()}
def _compute_loss(
self, z_log_var, z_mean, qed_true, qed_pred, graph_real, graph_generated
):
adjacency_real, features_real = graph_real
adjacency_gen, features_gen = graph_generated
adjacency_loss = tf.reduce_mean(
tf.reduce_sum(
keras.losses.categorical_crossentropy(adjacency_real, adjacency_gen),
axis=(1, 2),
)
)
features_loss = tf.reduce_mean(
tf.reduce_sum(
keras.losses.categorical_crossentropy(features_real, features_gen),
axis=(1),
)
)
kl_loss = -0.5 * tf.reduce_sum(
1 + z_log_var - tf.square(z_mean) - tf.exp(z_log_var), 1
)
kl_loss = tf.reduce_mean(kl_loss)
property_loss = tf.reduce_mean(
keras.losses.binary_crossentropy(qed_true, qed_pred)
)
graph_loss = self._gradient_penalty(graph_real, graph_generated)
return kl_loss + property_loss + graph_loss + adjacency_loss + features_loss
def _gradient_penalty(self, graph_real, graph_generated):
# 그래프 언팩
adjacency_real, features_real = graph_real
adjacency_generated, features_generated = graph_generated
# 보간된 그래프(adjacency_interp 및 features_interp) 생성
alpha = tf.random.uniform([self.batch_size])
alpha = tf.reshape(alpha, (self.batch_size, 1, 1, 1))
adjacency_interp = (adjacency_real * alpha) + (1 - alpha) * adjacency_generated
alpha = tf.reshape(alpha, (self.batch_size, 1, 1))
features_interp = (features_real * alpha) + (1 - alpha) * features_generated
# 보간된 그래프의 로짓 계산
with tf.GradientTape() as tape:
tape.watch(adjacency_interp)
tape.watch(features_interp)
_, _, logits, _, _ = self(
[adjacency_interp, features_interp], training=True
)
# 보간된 그래프에 대한 기울기 계산
grads = tape.gradient(logits, [adjacency_interp, features_interp])
# 기울기 패널티 계산
grads_adjacency_penalty = (1 - tf.norm(grads[0], axis=1)) ** 2
grads_features_penalty = (1 - tf.norm(grads[1], axis=2)) ** 2
return tf.reduce_mean(
tf.reduce_mean(grads_adjacency_penalty, axis=(-2, -1))
+ tf.reduce_mean(grads_features_penalty, axis=(-1))
)
def inference(self, batch_size):
z = tf.random.normal((batch_size, LATENT_DIM))
reconstruction_adjacency, reconstruction_features = model.decoder.predict(z)
# 인접 텐서에 대해 원-핫 인코딩 수행
adjacency = tf.argmax(reconstruction_adjacency, axis=1)
adjacency = tf.one_hot(adjacency, depth=BOND_DIM, axis=1)
# 인접 텐서에서 자가 결합 제거
adjacency = tf.linalg.set_diag(adjacency, tf.zeros(tf.shape(adjacency)[:-1]))
# 특성 텐서에 대해 원-핫 인코딩 수행
features = tf.argmax(reconstruction_features, axis=2)
features = tf.one_hot(features, depth=ATOM_DIM, axis=2)
return [
graph_to_molecule([adjacency[i].numpy(), features[i].numpy()])
for i in range(batch_size)
]
def call(self, inputs):
z_mean, log_var = self.encoder(inputs)
z = Sampling()([z_mean, log_var])
gen_adjacency, gen_features = self.decoder(z)
property_pred = self.property_prediction_layer(z_mean)
return z_mean, log_var, property_pred, gen_adjacency, gen_features모델 트레이닝
vae_optimizer = tf.keras.optimizers.Adam(learning_rate=VAE_LR)
encoder = get_encoder(
gconv_units=[9],
adjacency_shape=(BOND_DIM, NUM_ATOMS, NUM_ATOMS),
feature_shape=(NUM_ATOMS, ATOM_DIM),
latent_dim=LATENT_DIM,
dense_units=[512],
dropout_rate=0.0,
)
decoder = get_decoder(
dense_units=[128, 256, 512],
dropout_rate=0.2,
latent_dim=LATENT_DIM,
adjacency_shape=(BOND_DIM, NUM_ATOMS, NUM_ATOMS),
feature_shape=(NUM_ATOMS, ATOM_DIM),
)
model = MoleculeGenerator(encoder, decoder, MAX_MOLSIZE)
model.compile(vae_optimizer)
history = model.fit([adjacency_tensor, feature_tensor, qed_tensor], epochs=EPOCHS)결과
Epoch 1/10
250/250 [==============================] - 24s 84ms/step - loss: 68958.3946
Epoch 2/10
250/250 [==============================] - 20s 79ms/step - loss: 68819.8421
Epoch 3/10
250/250 [==============================] - 20s 79ms/step - loss: 68830.6720
Epoch 4/10
250/250 [==============================] - 20s 79ms/step - loss: 68816.1486
Epoch 5/10
250/250 [==============================] - 20s 79ms/step - loss: 68825.9977
Epoch 6/10
250/250 [==============================] - 19s 78ms/step - loss: 68818.0771
Epoch 7/10
250/250 [==============================] - 19s 77ms/step - loss: 68815.8525
Epoch 8/10
250/250 [==============================] - 20s 78ms/step - loss: 68820.5459
Epoch 9/10
250/250 [==============================] - 21s 83ms/step - loss: 68806.9465
Epoch 10/10
250/250 [==============================] - 21s 84ms/step - loss: 68805.9879추론
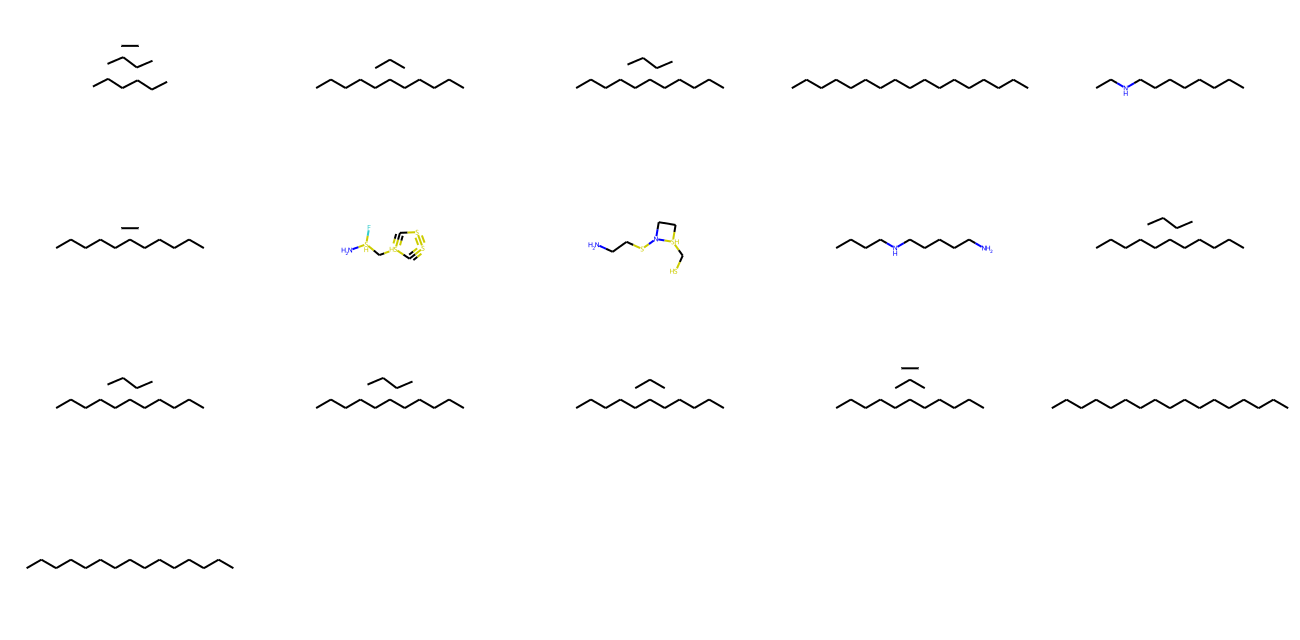
모델을 사용하여 잠재 공간의 다양한 지점에서 새로운 유효 분자를 생성합니다.
모델을 사용하여 unique 분자 생성
molecules = model.inference(1000)
MolsToGridImage(
[m for m in molecules if m is not None][:1000], molsPerRow=5, subImgSize=(260, 160)
)
분자 특성(QAE)에 따른 잠재 공간 클러스터 표시
def plot_latent(vae, data, labels):
# 잠재 공간에서 특성에 따른 2D 플롯을 표시합니다.
z_mean, _ = vae.encoder.predict(data)
plt.figure(figsize=(12, 10))
plt.scatter(z_mean[:, 0], z_mean[:, 1], c=labels)
plt.colorbar()
plt.xlabel("z[0]")
plt.ylabel("z[1]")
plt.show()
plot_latent(model, [adjacency_tensor[:8000], feature_tensor[:8000]], qed_tensor[:8000])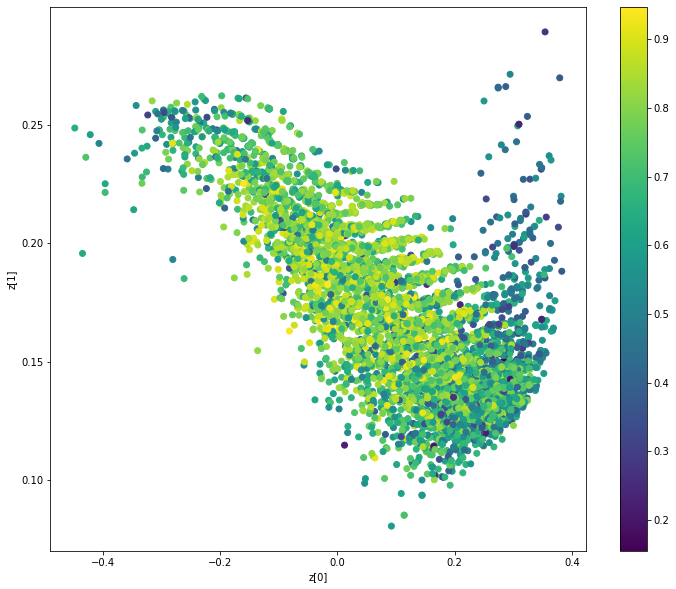
결론
이 예제에서는, 2016년의 “데이터 기반 연속 표현을 사용하는 자동 화학 설계” 논문과 2018년의 “MolGAN” 논문의 모델 아키텍처를 결합했습니다. 전자는 SMILES 입력을 문자열로 처리하여 SMILES 형식의 분자 문자열을 생성하려 하고, 후자는 SMILES 입력을 그래프(인접 행렬과 특성 행렬의 조합)로 고려하여 분자를 그래프로 생성하려 합니다.
이 하이브리드 접근 방식은 화학 공간을 탐색하는 새로운 유형의 유도된 경사 기반 검색을 가능하게 합니다.
예제는 HuggingFace에서 제공됩니다.
| 트레이닝된 모델 | 데모 |
|---|---|